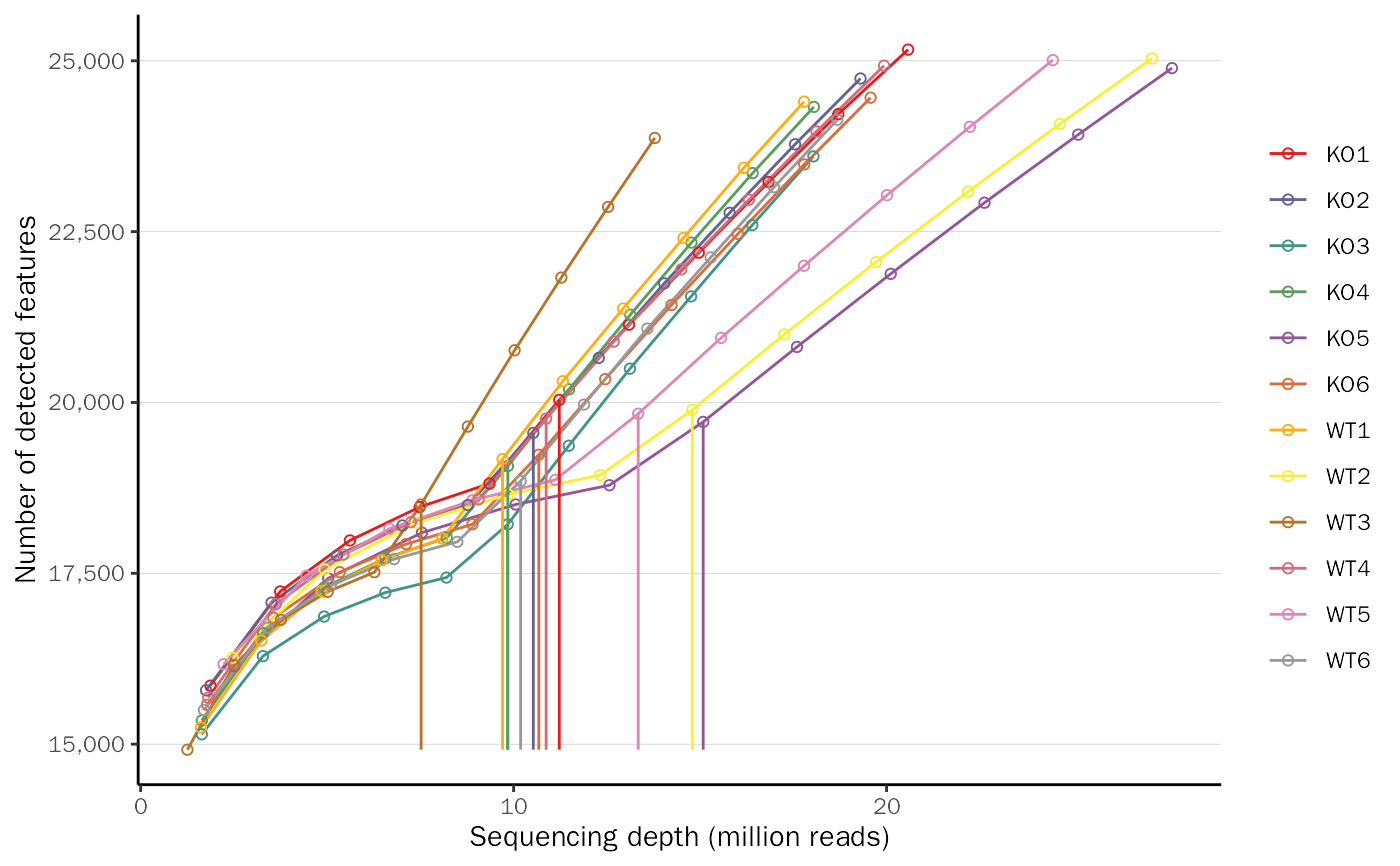
Count QC plot.
CountQC.RdCount QC plot.
CountQC( deobj, group.key = NULL, type = c("saturation", "cpm"), min.count = 0, ndepth = 10, cat.colors = NULL, ... )
Arguments
| deobj | Object created by DESeq2 or edgeR. |
|---|---|
| group.key | Sample group information. When set NULL, select first column of metadata. Default: NULL. |
| type | QC plot type, chosen from saturation and cpm. Default: saturation. |
| min.count | A feature is considered to be detected if the corresponding number of read counts is > |
| ndepth | Number of different sequencing depths to be simulated and plotted apart from the real depth. Default: 10. This parameter is only used by type "saturation". |
| cat.colors | Color used for different CPM Threshold groups or samples. Default: NULL (auto-selection). |
| ... | Parameters for |
Value
Count QC plot
Examples
library(DESeq2) library(DEbPeak) count.file <- system.file("extdata", "snon_count.txt", package = "DEbPeak") meta.file <- system.file("extdata", "snon_meta.txt", package = "DEbPeak") count.matrix <- read.table(file = count.file, header = TRUE, sep = "\t") meta.info <- read.table(file = meta.file, header = TRUE) dds <- DESeq2::DESeqDataSetFromMatrix(countData = count.matrix, colData = meta.info, design = ~condition)#> Warning: some variables in design formula are characters, converting to factorsCountQC(deobj = dds, group.key = "condition", type = "cpm")#>#> [1] "Warning: 25096 features with 0 counts in all samples are to be removed for this analysis." #> [1] "Count distributions are to be computed for:" #> [1] "KO1" "KO2" "KO3" "KO4" "KO5" "KO6" "WT1" "WT2" "WT3" "WT4" "WT5" "WT6"CountQC(deobj = dds, group.key = "condition", type = "saturation")#>