PrincipalComponentAnalysisPeak
PrincipalComponentAnalysisPeak.RmdIntroduction
Here, we will perfrom principal component analysis (PCA) on peak-related data (eg: ChIP-seq, ATAC-seq, m6a-seq et al.)
Example data
The data used here are from In vivo CD8+ T cell CRISPR screening reveals control by Fli1 in infection and cancer:
- RNA-seq data: the sgCtrl vs sgFli1 RNA sequencing at D8 Cl13 p.i., the raw data are stored in GSE149838
- ATAC-seq data: sgCtrl vs sgFli1 ATAC sequencing at D9 Cl13 p.i., the raw data are stored in GSE149836
Count matrix
The raw data:
# read the raw data
atac.counts = utils::read.table(file = "/home/songyabing/R/learn/tmp/DEbPeak/GSE149836_combUnionReadsWithLabels.txt",
sep = "\t", header = T)
head(atac.counts)## chr start end X290_1_S4 X290_2_S5 X360_1_S6 X360_2_S7 R_1_S2 R_2_S3
## 1 chr1 3131803 3131948 2 15 0 3 4 0
## 2 chr1 3210046 3210121 0 1 0 1 0 0
## 3 chr1 3265577 3265648 3 2 0 8 5 2
## 4 chr1 3345206 3345286 3 2 1 0 2 1
## 5 chr1 3505380 3505504 3 4 0 12 4 2
## 6 chr1 3513642 3513745 0 0 2 3 13 2To perform analysis (including Quality Control), we need to preprocess the data (row: feature, column: sample):
# read the processed data, the feature is consist of peak region, gene symbol and annotated binding region.
atac.counts.file = system.file("extdata", "RA_ATAC_count.txt", package = "DEbPeak")
atac.counts = utils::read.table(file = atac.counts.file, sep = "\t", header = T)
head(atac.counts)## Fli1KO_1 Fli1KO_2 Fli1KO_3
## chr1_GL456211_random:112828-112899|LOC100041034|DI 4 6 14
## chr1_GL456211_random:113055-113190|LOC100041034|DI 7 13 10
## chr1_GL456211_random:147874-147989|LOC100041034|DI 15 16 22
## chr1_GL456211_random:167241-167436|LOC100041034|DI 53 40 43
## chr1_GL456211_random:174677-174747|LOC100041034|I 9 6 17
## chr1_GL456211_random:199244-199444|LOC100041034|DI 23 14 26
## Fli1KO_4 WT_1 WT_2
## chr1_GL456211_random:112828-112899|LOC100041034|DI 12 10 3
## chr1_GL456211_random:113055-113190|LOC100041034|DI 11 21 31
## chr1_GL456211_random:147874-147989|LOC100041034|DI 14 23 27
## chr1_GL456211_random:167241-167436|LOC100041034|DI 54 39 46
## chr1_GL456211_random:174677-174747|LOC100041034|I 6 14 10
## chr1_GL456211_random:199244-199444|LOC100041034|DI 11 27 46If you have raw bam files of input/control and treatment samples and sample metadata, DEbPeak provides PeakMatrix to prepare the above count matrix.
Sample metadata
And, we also need the sample metadata:
# read the processed data
atac.meta.file = system.file("extdata", "RA_ATAC_meta.txt", package = "DEbPeak")
atac.meta = utils::read.table(file = atac.meta.file, sep = "\t", header = T)
head(atac.meta)## condition
## Fli1KO_1 KO
## Fli1KO_2 KO
## Fli1KO_3 KO
## Fli1KO_4 KO
## WT_1 WT
## WT_2 WTCreate DESeqDataSet
With above data, we can create DESeqDataSet object:
suppressWarnings(suppressMessages(library(DESeq2)))
# create dds
dds.atac <- DESeq2::DESeqDataSetFromMatrix(
countData = atac.counts, colData = atac.meta,
design = ~condition
)## Warning in DESeqDataSet(se, design = design, ignoreRank): some variables in
## design formula are characters, converting to factorsPCA
We will perform PCA with prcomp:
# conduct PCA
pca.res=PCA(deobj = dds.atac,transform.method = "rlog")
# get basic plots
basic.plots=PCABasic(pca.res,colby="condition",legend.pos = "right")Scree plot
A Scree Plot is a simple line plot that shows the total amount of variance that can be explained by each individual PC (Y-axis shows explained variance, X-axis shows the number of PCs). It can be used to determine the number of PCs to be explored for downstream analysis. Here, we created a cumulative scree plot based on PCAtools, the red dashed line represents 90% explained variance.
basic.plots[["screen"]]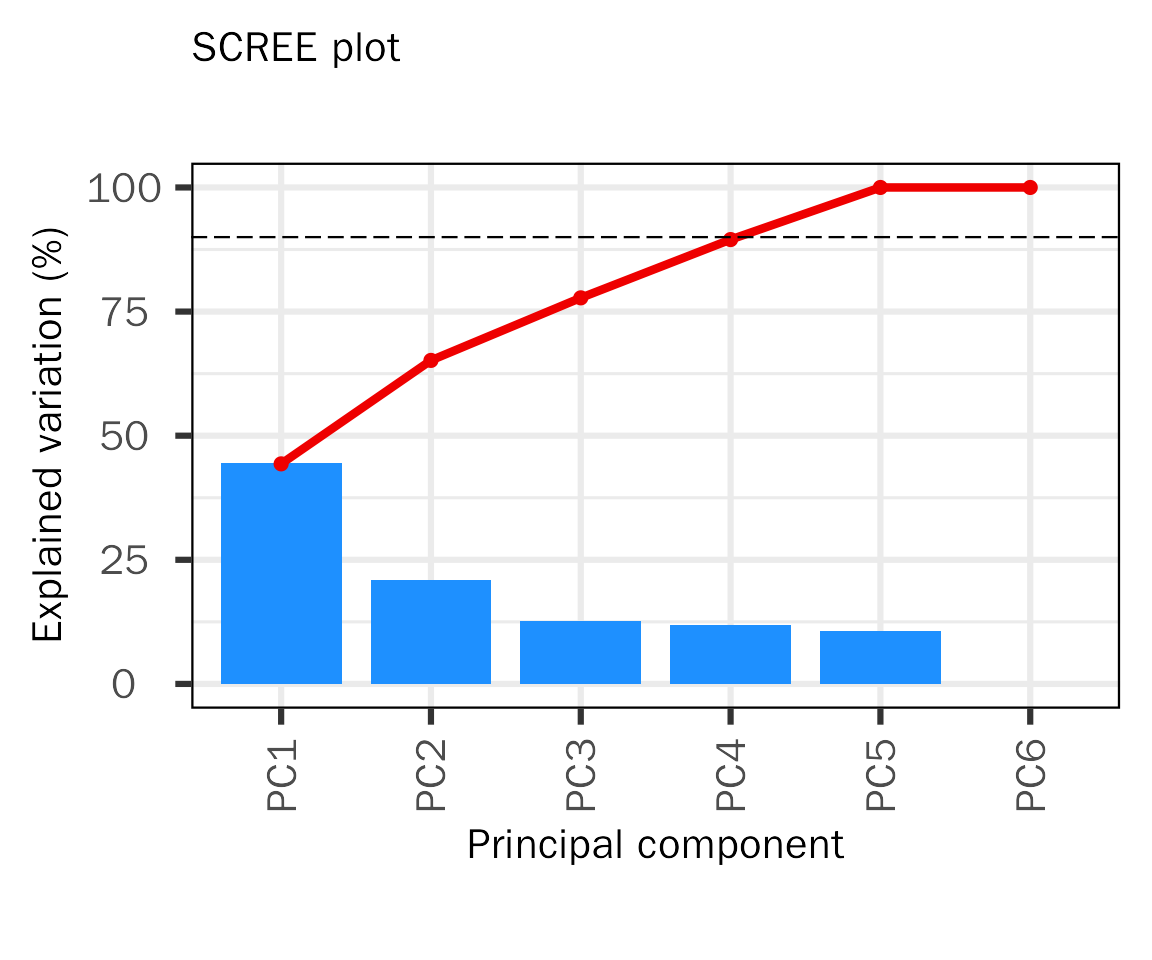
Biplot
Biplot contains informations from two aspects:
- sample similarity (point)
- how strongly each gene influences a principal component (vector)
- the vector will have projections on PCs, and the project values on each PC show how much weight they have on that PC. Positive values indicate that a vector/gene and a principal component are positively correlated whereas negative values indicate a negative correlation.
- the angle between vectors indicates how vectors/genes correlate with one another: a small angle implies positive correlation, a large one suggests negative correlation, and a 90 degree angle indicates no correlation between two vectors/genes.
The biplot is created based on PCAtools:
basic.plots[["biplot"]]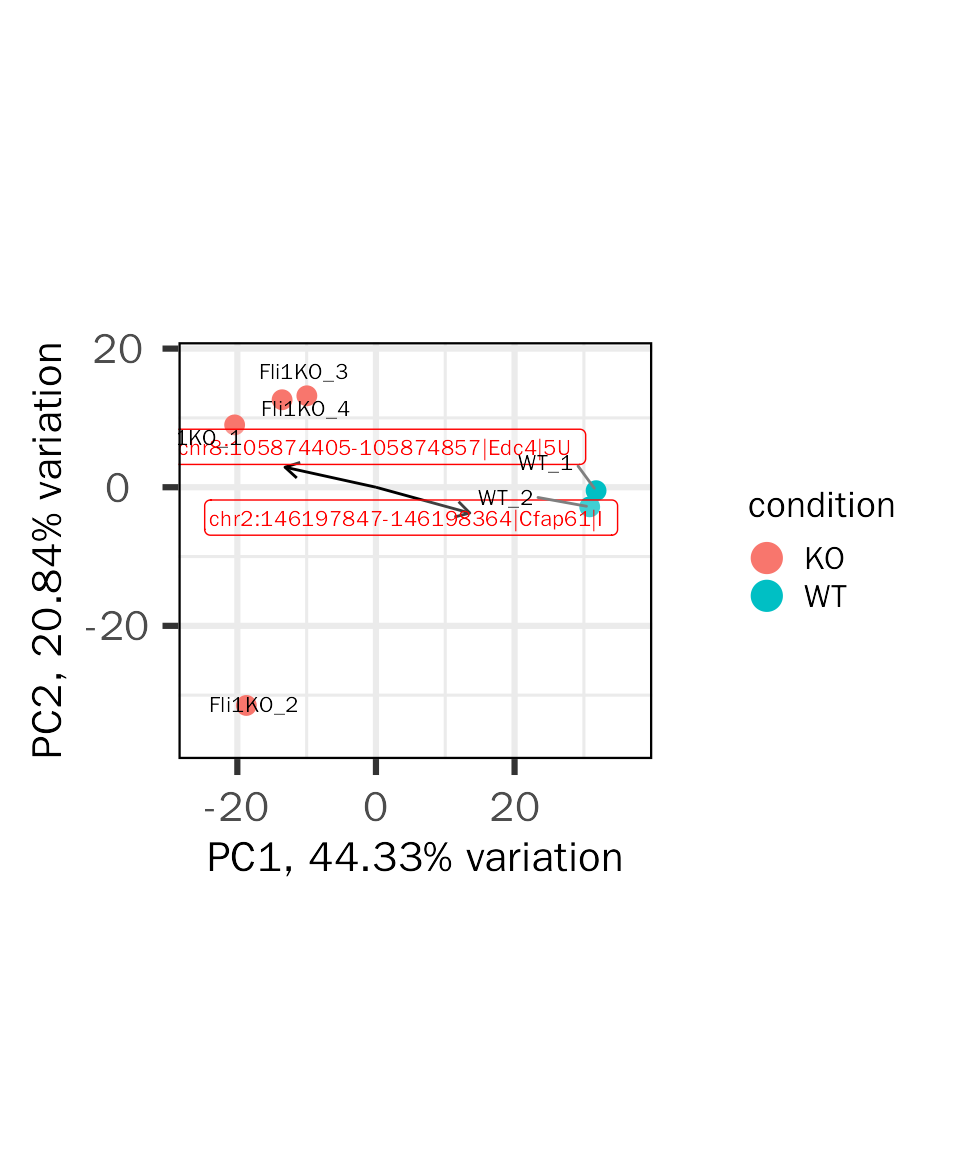
PC pairs plot
The PC pairs plot (based on PCAtools) will show sample similarity across different PC combination:
basic.plots[["pairs"]]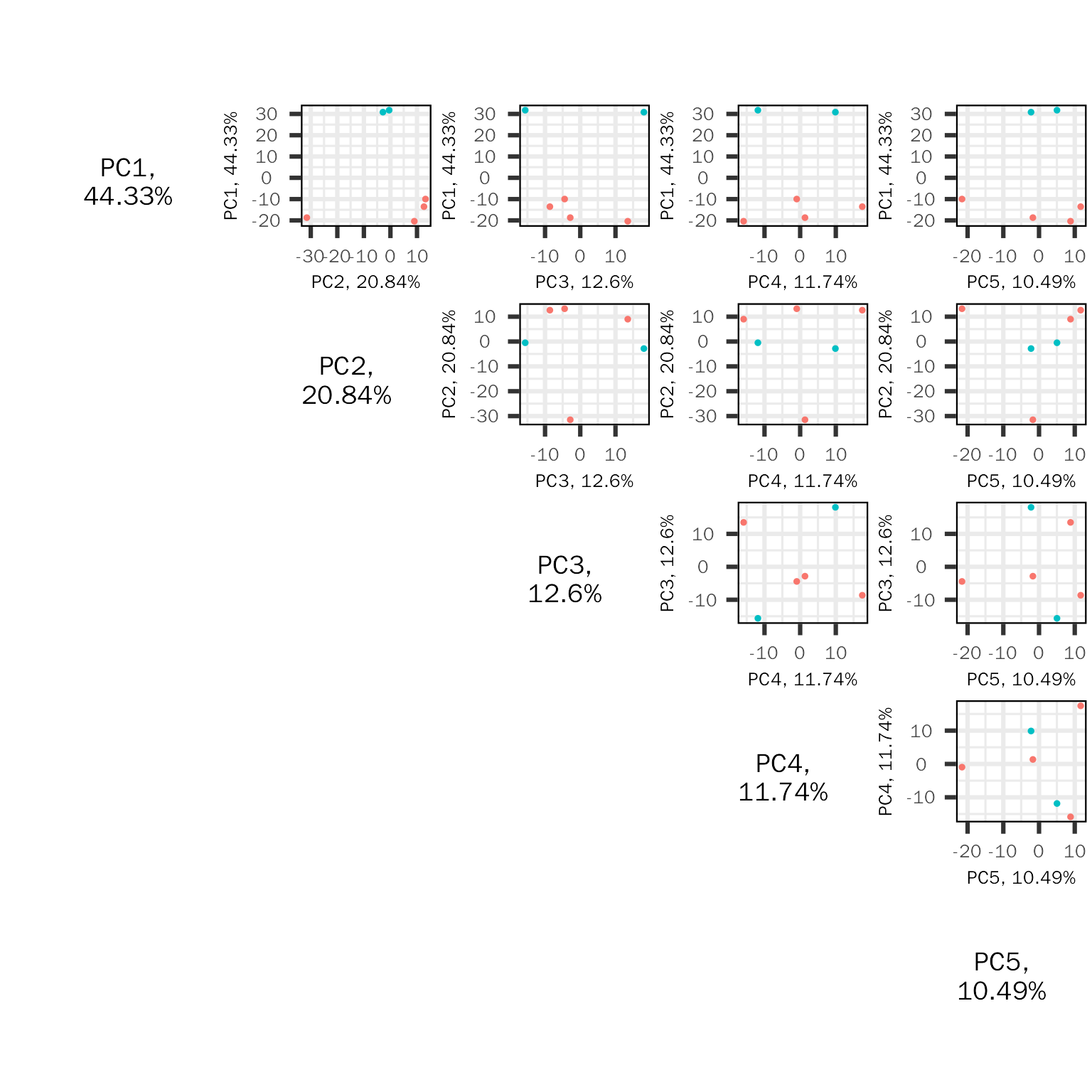
Lodding-related analysis
PCA loadings are the coefficients of the linear combination of the original variables from which the principal components (PCs) are constructed, which are simply the weights of the original variables. Here, lodding indicates how strongly each gene influences a principal component (Similar to vector in Biplot, but here we will evaluate each individual PC). Loadings range from -1 to 1, a high absolute value indicates that the gene strongly influences the component, and the sign of a loading indicates whether a whether a gene and a principal component are positively or negatively correlated.
The lodding-related analysis in DEbPeak contains four aspects:
- Loading bar plot: Visualize the genes with top n positive or negative loaddings of PCs
- Loading heatmap: Log2-normalized counts of the genes with top n positive or negative loaddings of PCs across samples
- Export genes: Export the genes with top n positive or negative loaddings of PCs
- GO enrichment: GO enrichment on the genes with top n positive or negative loaddings of PCs
Loading bar plot
Different from those in RNA-seq Principal Component Analysis (RNA-seq), we need to use data.type to indicate that the data are from peak-related data, and peak.anno.key to specify the peak binding to which region are used for analysis, available peak.anno.key values are: Promoter, 5' UTR, 3' UTR, Exon, Intron, Downstream, Distal Intergenic, All.
# loading bar plot
LoadingPlot(pca.res, data.type = "ATAC", peak.anno.key = "All", type = "bar")
Loading heatmap
# loading heatmap
LoadingPlot(pca.res, deobj = dds.atac, data.type = "ATAC", peak.anno.key = "All", type = "heat")
Export genes
# export loading genes on PC1 and PC2 (200 positive genes and 200 negative genes)
loading.gene.df=ExportPCGenes(pca = pca.res, data.type = "ATAC", peak.anno.key = "All", pc = 1:2, gene.num = 200)
head(loading.gene.df)## PC Gene Loadding Type
## 1 PC1 chr2:146197847-146198364|Cfap61|I 0.03426567 Positive
## 2 PC1 chrUn_JH584304:103171-103639|Pisd-ps3|DI 0.03166168 Positive
## 3 PC1 chrUn_JH584304:98078-98476|Pisd-ps3|DI 0.03154830 Positive
## 4 PC1 chrUn_JH584304:59463-60087|Pisd-ps3|P 0.03154772 Positive
## 5 PC1 chr13:74528968-74529954|Zfp825|DI 0.03092535 Positive
## 6 PC1 chr16:97401751-97402110|Bace2|I 0.03020087 PositiveGO enrichment
# save results to working folder
# LoadingGO(pca.res,gene.type="ENSEMBL",go.type="BP",padj.method="BH",save = T)
# return list
loading.go.results=LoadingGO(pca.res,gene.type="SYMBOL", data.type = "ATAC", peak.anno.key = "All",
go.type="BP",padj.method="BH",str.width = 50,save = F)Genes with positive loaddings
# positive loading genes
positive_go_results=loading.go.results[["Positive"]][["GO"]]
head(positive_go_results[["table"]])## [1] ID Description GeneRatio BgRatio pvalue p.adjust
## [7] qvalue geneID Count
## <0 行> (或0-长度的row.names)
positive_go_results[["plot"]]
Genes with negative loaddings
# negative loading genes
negative_go_results=loading.go.results[["Negative"]][["GO"]]
head(negative_go_results[["table"]])## [1] ID Description GeneRatio BgRatio pvalue p.adjust
## [7] qvalue geneID Count
## <0 行> (或0-长度的row.names)
negative_go_results[["plot"]]
3D visualization
To visualize three PCs simultaneously, DEbPeak provides PCA3D to create 3D PCA plot:
PCA3D(pca = pca.res, color.key = "condition", main = "3D PCA")
Session info
## R version 4.0.3 (2020-10-10)
## Platform: x86_64-conda-linux-gnu (64-bit)
## Running under: CentOS Linux 7 (Core)
##
## Matrix products: default
## BLAS/LAPACK: /home/softwares/anaconda3/envs/r4.0/lib/libopenblasp-r0.3.12.so
##
## locale:
## [1] LC_CTYPE=zh_CN.UTF-8 LC_NUMERIC=C
## [3] LC_TIME=zh_CN.UTF-8 LC_COLLATE=zh_CN.UTF-8
## [5] LC_MONETARY=zh_CN.UTF-8 LC_MESSAGES=zh_CN.UTF-8
## [7] LC_PAPER=zh_CN.UTF-8 LC_NAME=C
## [9] LC_ADDRESS=C LC_TELEPHONE=C
## [11] LC_MEASUREMENT=zh_CN.UTF-8 LC_IDENTIFICATION=C
##
## attached base packages:
## [1] stats4 stats graphics grDevices utils datasets methods
## [8] base
##
## other attached packages:
## [1] org.Mm.eg.db_3.12.0 AnnotationDbi_1.52.0
## [3] DEbPeak_1.4.0 DESeq2_1.30.1
## [5] SummarizedExperiment_1.20.0 Biobase_2.50.0
## [7] MatrixGenerics_1.2.1 matrixStats_0.58.0
## [9] GenomicRanges_1.42.0 GenomeInfoDb_1.26.7
## [11] IRanges_2.24.1 S4Vectors_0.28.1
## [13] BiocGenerics_0.42.0
##
## loaded via a namespace (and not attached):
## [1] rsvd_1.0.3
## [2] ggvenn_0.1.9
## [3] apeglm_1.12.0
## [4] Rsamtools_2.6.0
## [5] rsvg_2.1
## [6] foreach_1.5.1
## [7] rprojroot_2.0.2
## [8] crayon_1.4.1
## [9] V8_3.4.2
## [10] MASS_7.3-58
## [11] nlme_3.1-152
## [12] backports_1.2.1
## [13] sva_3.38.0
## [14] GOSemSim_2.25.0
## [15] rlang_1.1.0
## [16] XVector_0.30.0
## [17] readxl_1.4.2
## [18] irlba_2.3.5
## [19] limma_3.46.0
## [20] GOstats_2.56.0
## [21] BiocParallel_1.24.1
## [22] rjson_0.2.20
## [23] bit64_4.0.5
## [24] glue_1.6.2
## [25] DiffBind_3.0.15
## [26] mixsqp_0.3-43
## [27] pheatmap_1.0.12
## [28] parallel_4.0.3
## [29] DEFormats_1.18.0
## [30] base64url_1.4
## [31] tcltk_4.0.3
## [32] DOSE_3.23.2
## [33] haven_2.5.2
## [34] tidyselect_1.2.0
## [35] rio_0.5.27
## [36] XML_3.99-0.6
## [37] tidyr_1.3.0
## [38] ggpubr_0.4.0
## [39] GenomicAlignments_1.26.0
## [40] xtable_1.8-4
## [41] ggnetwork_0.5.12
## [42] magrittr_2.0.3
## [43] evaluate_0.14
## [44] ggplot2_3.4.2
## [45] cli_3.6.1
## [46] zlibbioc_1.36.0
## [47] hwriter_1.3.2
## [48] rstudioapi_0.14
## [49] bslib_0.3.1
## [50] GreyListChIP_1.22.0
## [51] fastmatch_1.1-3
## [52] BiocSingular_1.6.0
## [53] xfun_0.30
## [54] askpass_1.1
## [55] clue_0.3-59
## [56] gson_0.0.9
## [57] cluster_2.1.1
## [58] caTools_1.18.2
## [59] tidygraph_1.2.0
## [60] tibble_3.2.1
## [61] ggrepel_0.9.1
## [62] Biostrings_2.58.0
## [63] png_0.1-7
## [64] withr_2.5.0
## [65] bitops_1.0-6
## [66] ggforce_0.3.3
## [67] RBGL_1.66.0
## [68] plyr_1.8.6
## [69] cellranger_1.1.0
## [70] GSEABase_1.52.1
## [71] pcaPP_2.0-1
## [72] dqrng_0.2.1
## [73] coda_0.19-4
## [74] pillar_1.9.0
## [75] gplots_3.1.1
## [76] GlobalOptions_0.1.2
## [77] cachem_1.0.4
## [78] GenomicFeatures_1.42.2
## [79] fs_1.5.0
## [80] GetoptLong_1.0.5
## [81] clusterProfiler_4.7.1
## [82] DelayedMatrixStats_1.12.3
## [83] vctrs_0.6.2
## [84] generics_0.1.0
## [85] plot3D_1.4
## [86] tools_4.0.3
## [87] foreign_0.8-81
## [88] NOISeq_2.34.0
## [89] munsell_0.5.0
## [90] tweenr_1.0.2
## [91] fgsea_1.16.0
## [92] DelayedArray_0.16.3
## [93] fastmap_1.1.0
## [94] compiler_4.0.3
## [95] abind_1.4-5
## [96] rtracklayer_1.50.0
## [97] TxDb.Hsapiens.UCSC.hg19.knownGene_3.2.2
## [98] GenomeInfoDbData_1.2.4
## [99] gridExtra_2.3
## [100] edgeR_3.32.1
## [101] lattice_0.20-45
## [102] ggnewscale_0.4.7
## [103] AnnotationForge_1.32.0
## [104] utf8_1.2.1
## [105] dplyr_1.1.2
## [106] BiocFileCache_1.14.0
## [107] jsonlite_1.8.4
## [108] scales_1.2.1
## [109] graph_1.68.0
## [110] carData_3.0-4
## [111] sparseMatrixStats_1.2.1
## [112] TFEA.ChIP_1.10.0
## [113] genefilter_1.72.1
## [114] car_3.0-11
## [115] doParallel_1.0.16
## [116] latticeExtra_0.6-29
## [117] R.utils_2.12.0
## [118] brew_1.0-6
## [119] checkmate_2.0.0
## [120] rmarkdown_2.14
## [121] openxlsx_4.2.3
## [122] pkgdown_1.6.1
## [123] cowplot_1.1.1
## [124] textshaping_0.3.6
## [125] forcats_1.0.0
## [126] downloader_0.4
## [127] BSgenome_1.58.0
## [128] igraph_1.4.99.9024
## [129] survival_3.2-10
## [130] numDeriv_2016.8-1.1
## [131] yaml_2.2.1
## [132] plotrix_3.8-2
## [133] systemfonts_1.0.4
## [134] ashr_2.2-47
## [135] SQUAREM_2021.1
## [136] htmltools_0.5.2
## [137] memoise_2.0.0
## [138] VariantAnnotation_1.36.0
## [139] locfit_1.5-9.4
## [140] graphlayouts_0.7.1
## [141] batchtools_0.9.15
## [142] PCAtools_2.2.0
## [143] viridisLite_0.4.0
## [144] rrcov_1.7-0
## [145] digest_0.6.27
## [146] assertthat_0.2.1
## [147] rappdirs_0.3.3
## [148] emdbook_1.3.12
## [149] RSQLite_2.2.5
## [150] amap_0.8-18
## [151] yulab.utils_0.0.4
## [152] debugme_1.1.0
## [153] misc3d_0.9-1
## [154] data.table_1.14.2
## [155] blob_1.2.1
## [156] R.oo_1.24.0
## [157] ragg_0.4.0
## [158] labeling_0.4.2
## [159] splines_4.0.3
## [160] Cairo_1.5-12.2
## [161] ggupset_0.3.0
## [162] RCurl_1.98-1.3
## [163] broom_1.0.4
## [164] hms_1.1.3
## [165] colorspace_2.0-0
## [166] BiocManager_1.30.16
## [167] shape_1.4.6
## [168] sass_0.4.1
## [169] GEOquery_2.58.0
## [170] Rcpp_1.0.9
## [171] mvtnorm_1.1-2
## [172] circlize_0.4.15
## [173] enrichplot_1.10.2
## [174] fansi_0.4.2
## [175] tzdb_0.3.0
## [176] truncnorm_1.0-8
## [177] ChIPseeker_1.33.0.900
## [178] R6_2.5.0
## [179] grid_4.0.3
## [180] lifecycle_1.0.3
## [181] ShortRead_1.48.0
## [182] zip_2.1.1
## [183] curl_4.3
## [184] ggsignif_0.6.3
## [185] jquerylib_0.1.3
## [186] robustbase_0.95-0
## [187] DO.db_2.9
## [188] Matrix_1.5-4
## [189] qvalue_2.22.0
## [190] desc_1.3.0
## [191] org.Hs.eg.db_3.12.0
## [192] RColorBrewer_1.1-2
## [193] iterators_1.0.13
## [194] stringr_1.5.0
## [195] DOT_0.1
## [196] ggpie_0.2.5
## [197] beachmat_2.6.4
## [198] polyclip_1.10-0
## [199] biomaRt_2.46.3
## [200] purrr_1.0.1
## [201] shadowtext_0.0.9
## [202] gridGraphics_0.5-1
## [203] mgcv_1.8-34
## [204] ComplexHeatmap_2.13.1
## [205] openssl_1.4.3
## [206] patchwork_1.0.0
## [207] bdsmatrix_1.3-4
## [208] codetools_0.2-18
## [209] invgamma_1.1
## [210] GO.db_3.12.1
## [211] gtools_3.8.2
## [212] prettyunits_1.1.1
## [213] dbplyr_2.3.2
## [214] R.methodsS3_1.8.1
## [215] gtable_0.3.0
## [216] DBI_1.1.1
## [217] highr_0.8
## [218] ggfun_0.0.6
## [219] httr_1.4.5
## [220] KernSmooth_2.23-18
## [221] stringi_1.5.3
## [222] progress_1.2.2
## [223] reshape2_1.4.4
## [224] farver_2.1.0
## [225] annotate_1.68.0
## [226] viridis_0.6.1
## [227] Rgraphviz_2.34.0
## [228] xml2_1.3.4
## [229] bbmle_1.0.24
## [230] systemPipeR_1.24.3
## [231] boot_1.3-28
## [232] readr_2.1.4
## [233] geneplotter_1.68.0
## [234] ggplotify_0.1.0
## [235] Category_2.56.0
## [236] DEoptimR_1.0-11
## [237] bit_4.0.4
## [238] scatterpie_0.1.7
## [239] jpeg_0.1-8.1
## [240] ggraph_2.0.5
## [241] pkgconfig_2.0.3
## [242] rstatix_0.7.0
## [243] knitr_1.37