IntegrateChIPATAC
IntegrateChIPATAC.RmdIntroduction
In previous vignettes, we have introduced intregrating RNA-seq with ATAC-seq and ChIP-seq separately. But we should keep in mind that ATAC-seq and ChIP-seq are highly consistent and complementary. Most TFs require open chromatin for binding to DNA recognition sites, ATAC-seq peaks generally overlap with TF ChIP-seq peaks but are often broader. Thus, TF ChIP-seq and ATAC-seq can mutually validate the quality and reliability of each other within the same experimental system.
To obtain more accurate gene regulation results, here we will integrate RNA-seq with ChIP-seq and ATAC-seq simultaneously.
Example data
The data used here contains RNA-seq and ChIP-seq datasets from RUNX represses Pmp22 to drive neurofibromagenesis:
- RNA-seq: two genotypes and three samples per genotype, the raw data are stored in GSE122774
- ChIP-seq: two genotypes and one sample per genotype, the raw data are stored in GSE122775
- ATAC-seq: two genotypes and one sample per genotype, the raw data are stored in GSE122776
Load results
The integration is based on results of “IntegrateChIP” vignette and “IntegrateATAC” vignette, load the results first.
# library
suppressWarnings(suppressMessages(library(DESeq2)))
suppressWarnings(suppressMessages(library(DEbPeak)))
# load RNA-seq and ChIP-seq integration results
load(file = "/home/songyabing/R/learn/tmp/DEbPeak/RNAandChIP.RData")
# load RNA-seq and ATAC-seq integration results
load(file = "/home/songyabing/R/learn/tmp/DEbPeak/RNAandATAC.RData")Integrate RNA-seq, ChIP-seq and ATAC-seq
Integrate
debpeak.res = DEbCA(de.res = debchip.dds.results.ordered, chip.peak.res = peak.anno.df,
atac.peak.res = atac.peak.anno.df,
peak.anno.key = "Promoter", merge.key = "SYMBOL")## Differential expression analysis with DESeq2!
head(debpeak.res)## SYMBOL geneId_ChIP Peak_ChIP annotation_ChIP
## 1 0610009E02Rik <NA> <NA> <NA>
## 2 0610012G03Rik <NA> <NA> <NA>
## 3 0610039K10Rik <NA> <NA> <NA>
## 4 1110002J07Rik 68488 chr10:66905070-66905215 Promoter (12-13kb)
## 5 1110004F10Rik <NA> <NA> <NA>
## 6 1110008P14Rik <NA> <NA> <NA>
## anno_ChIP ENSEMBL_ChIP GENENAME_ChIP log2FoldChange abundance
## 1 <NA> <NA> <NA> NA NA
## 2 <NA> <NA> <NA> 1.837668 59.17919
## 3 <NA> <NA> <NA> NA NA
## 4 Promoter <NA> RIKEN cDNA 1110002J07 gene NA NA
## 5 <NA> <NA> <NA> NA NA
## 6 <NA> <NA> <NA> 1.013546 125.76554
## signif regulation Type1 geneId_ATAC Peak_ATAC
## 1 NA <NA> <NA> 100125929 chr2:26446213-26446286
## 2 3.073773 Up_regulated UP 106264 chr16:31948207-31948280
## 3 NA <NA> <NA> 68386 chr2:163644811-163644884
## 4 NA <NA> ChIP 68488 chr10:66935911-66935984
## 5 NA <NA> <NA> 56372 chr7:116039769-116039842
## 6 2.277566 Up_regulated UP <NA> <NA>
## annotation_ATAC anno_ATAC ENSEMBL_ATAC GENENAME_ATAC
## 1 Promoter (<=1kb) Promoter ENSMUSG00000086714 RIKEN cDNA 0610009E02 gene
## 2 Promoter (<=1kb) Promoter ENSMUSG00000107002 RIKEN cDNA 0610012G03 gene
## 3 Promoter (<=1kb) Promoter ENSMUSG00000058812 RIKEN cDNA 0610039K10 gene
## 4 Promoter (15-16kb) Promoter <NA> RIKEN cDNA 1110002J07 gene
## 5 Promoter (<=1kb) Promoter ENSMUSG00000030663 RIKEN cDNA 1110004F10 gene
## 6 <NA> <NA> <NA> <NA>
## Type geneId
## 1 ATAC 100125929
## 2 UPbATAC 106264
## 3 ATAC 68386
## 4 ChIPbATAC 68488
## 5 ATAC 56372
## 6 UP <NA>Integrate summary
# DE and ChIP venn plot
# debpeak.plot = PlotDEbPeak(debpeak.res, peak.type = "Peak", show_percentage=FALSE)
# debpeak.plot
debpeak.plot = InteVenn(inte.res = debpeak.res, inte.type = "DEbPeak", peak.type = "Peak",
gene.col = "SYMBOL",show_percentage = FALSE)
debpeak.plot
Functional enrichment
There are five categories for users to choose to perform functional enrichment: ChIP, ATAC, UP, DOWN, ChIPbATAC, UPbChIP, DOWNbChIP, UPbPeak, DOWNbPeak, UPbATAC, DOWNbATAC. Here, we will use DOWNbPeak and UPbPeak as examples.
UPbPeak
# functional enrichment on direct targets
# debpeak.up.fe.results = DEbPeakFE(de.peak = debpeak.res, peak.fe.key = "UPbPeak", gene.type = "ENTREZID", species="Mouse",save = F)
debpeak.up.fe.results = InteFE(inte.res = debpeak.res, fe.key = "UPbPeak", inte.type = "DEbPeak",
gene.type = "ENTREZID", species="Mouse",save = F)## ## conduct ALL GO enrichment analysis on: UPbPeak## wrong orderBy parameter; set to default `orderBy = "x"`## Scale for y is already present.
## Adding another scale for y, which will replace the existing scale.
## wrong orderBy parameter; set to default `orderBy = "x"`
##
## Scale for y is already present.
## Adding another scale for y, which will replace the existing scale.The results:
# the result table
head(debpeak.up.fe.results[["GO"]][["table"]])## ONTOLOGY ID Description GeneRatio
## GO:0043218 CC GO:0043218 compact myelin 2/9
## GO:0043209 CC GO:0043209 myelin sheath 2/9
## GO:0070382 CC GO:0070382 exocytic vesicle 2/9
## GO:0043256 CC GO:0043256 laminin complex 1/9
## GO:0019898 CC GO:0019898 extrinsic component of membrane 2/9
## GO:0030133 CC GO:0030133 transport vesicle 2/9
## BgRatio pvalue p.adjust qvalue geneID Count
## GO:0043218 19/23271 2.265877e-05 0.001087621 0.000500878 Mbp/Pmp22 2
## GO:0043209 213/23271 2.877487e-03 0.043690148 0.020120463 Mbp/Pmp22 2
## GO:0070382 261/23271 4.282761e-03 0.043690148 0.020120463 Bsn/Syt6 2
## GO:0043256 12/23271 4.632203e-03 0.043690148 0.020120463 Pmp22 1
## GO:0019898 320/23271 6.366508e-03 0.043690148 0.020120463 Bsn/Syt6 2
## GO:0030133 339/23271 7.118989e-03 0.043690148 0.020120463 Bsn/Syt6 2
# the result plot
debpeak.up.fe.results[["GO"]][["plot"]]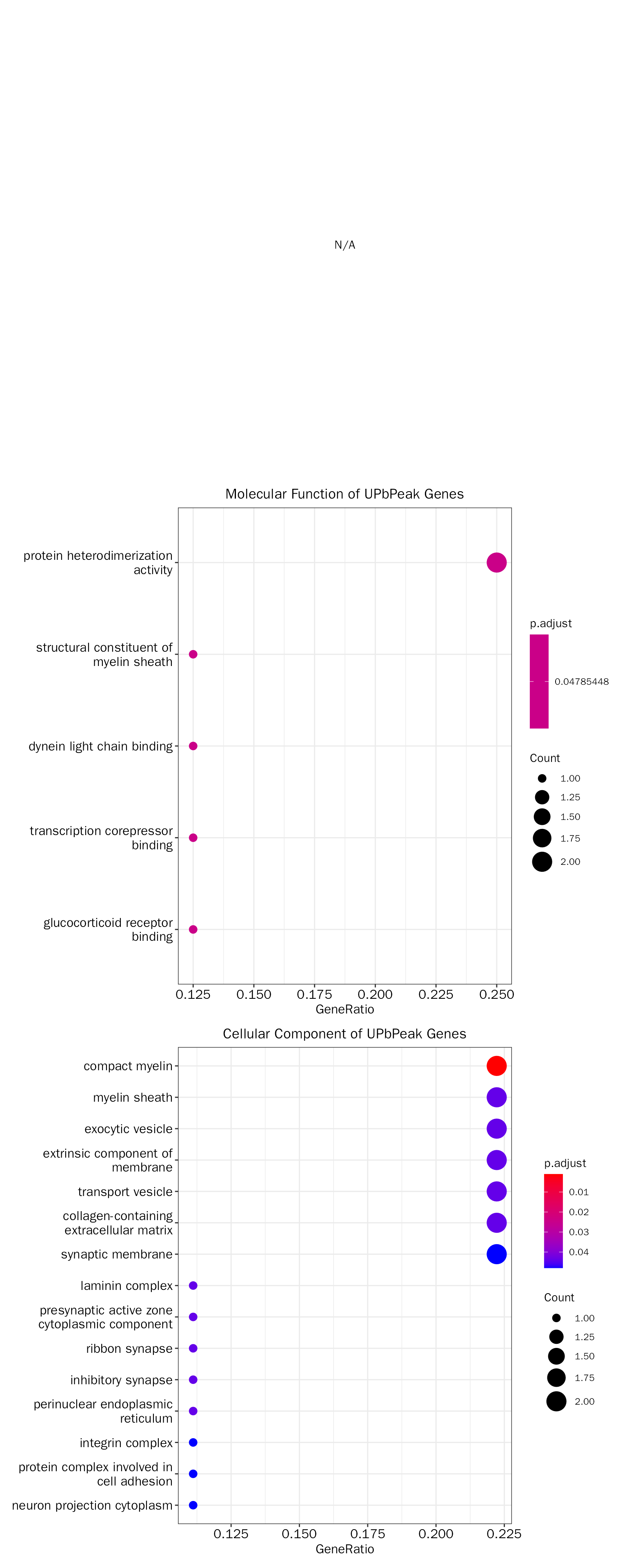
DOWNbPeak
# functional enrichment on direct targets
# debpeak.down.fe.results = DEbPeakFE(de.peak = debpeak.res, peak.fe.key = "DOWNbPeak",
# gene.type = "ENTREZID", species="Mouse",save = F)
debpeak.down.fe.results = InteFE(inte.res = debpeak.res, fe.key = "DOWNbPeak", inte.type = "DEbPeak",
gene.type = "ENTREZID", species="Mouse",save = F)## conduct ALL GO enrichment analysis on: DOWNbPeak## wrong orderBy parameter; set to default `orderBy = "x"`## Scale for y is already present.
## Adding another scale for y, which will replace the existing scale.
## wrong orderBy parameter; set to default `orderBy = "x"`
##
## Scale for y is already present.
## Adding another scale for y, which will replace the existing scale.The results:
# the result table
head(debpeak.down.fe.results[["GO"]][["table"]])## ONTOLOGY ID Description
## GO:0045064 BP GO:0045064 T-helper 2 cell differentiation
## GO:0010811 BP GO:0010811 positive regulation of cell-substrate adhesion
## GO:0045785 BP GO:0045785 positive regulation of cell adhesion
## GO:0045860 BP GO:0045860 positive regulation of protein kinase activity
## GO:0042832 BP GO:0042832 defense response to protozoan
## GO:0010810 BP GO:0010810 regulation of cell-substrate adhesion
## GeneRatio BgRatio pvalue p.adjust qvalue
## GO:0045064 2/13 17/23328 3.880421e-05 0.01212719 0.006013313
## GO:0010811 3/13 128/23328 4.433038e-05 0.01212719 0.006013313
## GO:0045785 4/13 435/23328 7.460788e-05 0.01212719 0.006013313
## GO:0045860 4/13 440/23328 7.798836e-05 0.01212719 0.006013313
## GO:0042832 2/13 36/23328 1.786843e-04 0.01717888 0.008518210
## GO:0010810 3/13 214/23328 2.034275e-04 0.01717888 0.008518210
## geneID Count
## GO:0045064 Bcl3/Il4ra 2
## GO:0010811 Col8a1/Fbln2/Sdc4 3
## GO:0045785 Col8a1/Fbln2/Il4ra/Sdc4 4
## GO:0045860 Akap13/Ccnd1/Ern1/Sdc4 4
## GO:0042832 Bcl3/Il4ra 2
## GO:0010810 Col8a1/Fbln2/Sdc4 3
# the result plot
debpeak.down.fe.results[["GO"]][["plot"]]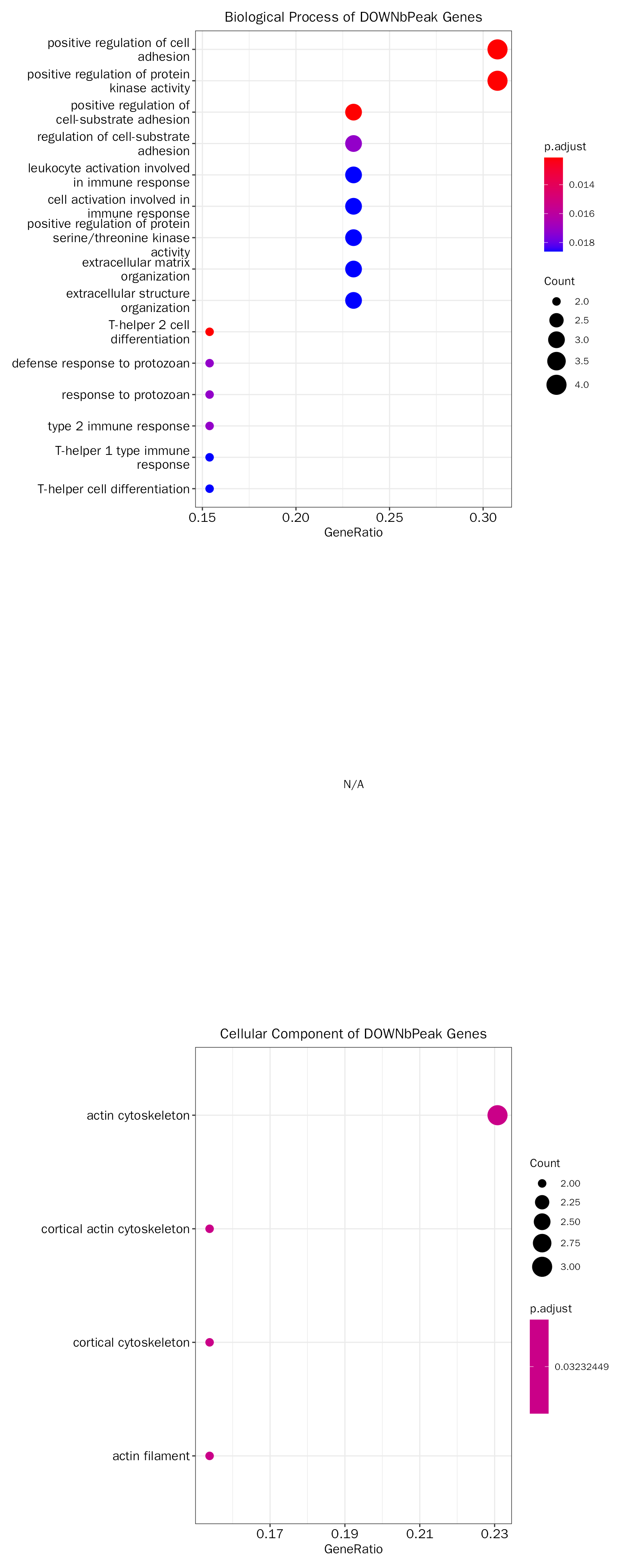
Session info
## R version 4.0.3 (2020-10-10)
## Platform: x86_64-conda-linux-gnu (64-bit)
## Running under: CentOS Linux 7 (Core)
##
## Matrix products: default
## BLAS/LAPACK: /home/softwares/anaconda3/envs/r4.0/lib/libopenblasp-r0.3.12.so
##
## locale:
## [1] LC_CTYPE=zh_CN.UTF-8 LC_NUMERIC=C
## [3] LC_TIME=zh_CN.UTF-8 LC_COLLATE=zh_CN.UTF-8
## [5] LC_MONETARY=zh_CN.UTF-8 LC_MESSAGES=zh_CN.UTF-8
## [7] LC_PAPER=zh_CN.UTF-8 LC_NAME=C
## [9] LC_ADDRESS=C LC_TELEPHONE=C
## [11] LC_MEASUREMENT=zh_CN.UTF-8 LC_IDENTIFICATION=C
##
## attached base packages:
## [1] stats4 stats graphics grDevices utils datasets methods
## [8] base
##
## other attached packages:
## [1] org.Mm.eg.db_3.12.0 AnnotationDbi_1.52.0
## [3] DEbPeak_1.4.0 DESeq2_1.30.1
## [5] SummarizedExperiment_1.20.0 Biobase_2.50.0
## [7] MatrixGenerics_1.2.1 matrixStats_0.58.0
## [9] GenomicRanges_1.42.0 GenomeInfoDb_1.26.7
## [11] IRanges_2.24.1 S4Vectors_0.28.1
## [13] BiocGenerics_0.42.0
##
## loaded via a namespace (and not attached):
## [1] rsvd_1.0.3
## [2] ggvenn_0.1.9
## [3] apeglm_1.12.0
## [4] Rsamtools_2.6.0
## [5] rsvg_2.1
## [6] foreach_1.5.1
## [7] rprojroot_2.0.2
## [8] crayon_1.4.1
## [9] V8_3.4.2
## [10] MASS_7.3-58
## [11] nlme_3.1-152
## [12] backports_1.2.1
## [13] sva_3.38.0
## [14] GOSemSim_2.25.0
## [15] rlang_1.1.0
## [16] XVector_0.30.0
## [17] readxl_1.4.2
## [18] irlba_2.3.5
## [19] limma_3.46.0
## [20] GOstats_2.56.0
## [21] BiocParallel_1.24.1
## [22] rjson_0.2.20
## [23] bit64_4.0.5
## [24] glue_1.6.2
## [25] DiffBind_3.0.15
## [26] mixsqp_0.3-43
## [27] pheatmap_1.0.12
## [28] parallel_4.0.3
## [29] DEFormats_1.18.0
## [30] base64url_1.4
## [31] tcltk_4.0.3
## [32] DOSE_3.23.2
## [33] haven_2.5.2
## [34] tidyselect_1.2.0
## [35] rio_0.5.27
## [36] XML_3.99-0.6
## [37] tidyr_1.3.0
## [38] ggpubr_0.4.0
## [39] GenomicAlignments_1.26.0
## [40] xtable_1.8-4
## [41] ggnetwork_0.5.12
## [42] magrittr_2.0.3
## [43] evaluate_0.14
## [44] ggplot2_3.4.2
## [45] cli_3.6.1
## [46] zlibbioc_1.36.0
## [47] hwriter_1.3.2
## [48] rstudioapi_0.14
## [49] bslib_0.3.1
## [50] GreyListChIP_1.22.0
## [51] fastmatch_1.1-3
## [52] BiocSingular_1.6.0
## [53] xfun_0.30
## [54] askpass_1.1
## [55] clue_0.3-59
## [56] gson_0.0.9
## [57] cluster_2.1.1
## [58] caTools_1.18.2
## [59] tidygraph_1.2.0
## [60] tibble_3.2.1
## [61] ggrepel_0.9.1
## [62] Biostrings_2.58.0
## [63] png_0.1-7
## [64] withr_2.5.0
## [65] bitops_1.0-6
## [66] ggforce_0.3.3
## [67] RBGL_1.66.0
## [68] plyr_1.8.6
## [69] cellranger_1.1.0
## [70] GSEABase_1.52.1
## [71] pcaPP_2.0-1
## [72] dqrng_0.2.1
## [73] coda_0.19-4
## [74] pillar_1.9.0
## [75] gplots_3.1.1
## [76] GlobalOptions_0.1.2
## [77] cachem_1.0.4
## [78] GenomicFeatures_1.42.2
## [79] fs_1.5.0
## [80] GetoptLong_1.0.5
## [81] clusterProfiler_4.7.1
## [82] DelayedMatrixStats_1.12.3
## [83] vctrs_0.6.2
## [84] generics_0.1.0
## [85] plot3D_1.4
## [86] tools_4.0.3
## [87] foreign_0.8-81
## [88] NOISeq_2.34.0
## [89] munsell_0.5.0
## [90] tweenr_1.0.2
## [91] fgsea_1.16.0
## [92] DelayedArray_0.16.3
## [93] fastmap_1.1.0
## [94] compiler_4.0.3
## [95] abind_1.4-5
## [96] rtracklayer_1.50.0
## [97] TxDb.Hsapiens.UCSC.hg19.knownGene_3.2.2
## [98] GenomeInfoDbData_1.2.4
## [99] gridExtra_2.3
## [100] edgeR_3.32.1
## [101] lattice_0.20-45
## [102] ggnewscale_0.4.7
## [103] AnnotationForge_1.32.0
## [104] utf8_1.2.1
## [105] dplyr_1.1.2
## [106] BiocFileCache_1.14.0
## [107] jsonlite_1.8.4
## [108] scales_1.2.1
## [109] graph_1.68.0
## [110] carData_3.0-4
## [111] sparseMatrixStats_1.2.1
## [112] TFEA.ChIP_1.10.0
## [113] genefilter_1.72.1
## [114] car_3.0-11
## [115] doParallel_1.0.16
## [116] latticeExtra_0.6-29
## [117] R.utils_2.12.0
## [118] brew_1.0-6
## [119] checkmate_2.0.0
## [120] rmarkdown_2.14
## [121] openxlsx_4.2.3
## [122] pkgdown_1.6.1
## [123] cowplot_1.1.1
## [124] textshaping_0.3.6
## [125] forcats_1.0.0
## [126] downloader_0.4
## [127] BSgenome_1.58.0
## [128] igraph_1.4.99.9024
## [129] survival_3.2-10
## [130] numDeriv_2016.8-1.1
## [131] yaml_2.2.1
## [132] plotrix_3.8-2
## [133] systemfonts_1.0.4
## [134] ashr_2.2-47
## [135] SQUAREM_2021.1
## [136] htmltools_0.5.2
## [137] memoise_2.0.0
## [138] VariantAnnotation_1.36.0
## [139] locfit_1.5-9.4
## [140] graphlayouts_0.7.1
## [141] batchtools_0.9.15
## [142] PCAtools_2.2.0
## [143] viridisLite_0.4.0
## [144] rrcov_1.7-0
## [145] digest_0.6.27
## [146] assertthat_0.2.1
## [147] rappdirs_0.3.3
## [148] emdbook_1.3.12
## [149] RSQLite_2.2.5
## [150] amap_0.8-18
## [151] yulab.utils_0.0.4
## [152] debugme_1.1.0
## [153] misc3d_0.9-1
## [154] data.table_1.14.2
## [155] blob_1.2.1
## [156] R.oo_1.24.0
## [157] ragg_0.4.0
## [158] labeling_0.4.2
## [159] splines_4.0.3
## [160] ggupset_0.3.0
## [161] RCurl_1.98-1.3
## [162] broom_1.0.4
## [163] hms_1.1.3
## [164] colorspace_2.0-0
## [165] BiocManager_1.30.16
## [166] shape_1.4.6
## [167] sass_0.4.1
## [168] GEOquery_2.58.0
## [169] Rcpp_1.0.9
## [170] mvtnorm_1.1-2
## [171] circlize_0.4.15
## [172] enrichplot_1.10.2
## [173] fansi_0.4.2
## [174] tzdb_0.3.0
## [175] truncnorm_1.0-8
## [176] ChIPseeker_1.33.0.900
## [177] R6_2.5.0
## [178] grid_4.0.3
## [179] lifecycle_1.0.3
## [180] ShortRead_1.48.0
## [181] zip_2.1.1
## [182] curl_4.3
## [183] ggsignif_0.6.3
## [184] jquerylib_0.1.3
## [185] robustbase_0.95-0
## [186] DO.db_2.9
## [187] Matrix_1.5-4
## [188] qvalue_2.22.0
## [189] desc_1.3.0
## [190] org.Hs.eg.db_3.12.0
## [191] RColorBrewer_1.1-2
## [192] iterators_1.0.13
## [193] stringr_1.5.0
## [194] DOT_0.1
## [195] ggpie_0.2.5
## [196] beachmat_2.6.4
## [197] polyclip_1.10-0
## [198] biomaRt_2.46.3
## [199] purrr_1.0.1
## [200] shadowtext_0.0.9
## [201] gridGraphics_0.5-1
## [202] mgcv_1.8-34
## [203] ComplexHeatmap_2.13.1
## [204] openssl_1.4.3
## [205] patchwork_1.0.0
## [206] bdsmatrix_1.3-4
## [207] codetools_0.2-18
## [208] invgamma_1.1
## [209] GO.db_3.12.1
## [210] gtools_3.8.2
## [211] prettyunits_1.1.1
## [212] dbplyr_2.3.2
## [213] R.methodsS3_1.8.1
## [214] gtable_0.3.0
## [215] DBI_1.1.1
## [216] highr_0.8
## [217] ggfun_0.0.6
## [218] httr_1.4.5
## [219] KernSmooth_2.23-18
## [220] stringi_1.5.3
## [221] progress_1.2.2
## [222] reshape2_1.4.4
## [223] farver_2.1.0
## [224] annotate_1.68.0
## [225] viridis_0.6.1
## [226] Rgraphviz_2.34.0
## [227] xml2_1.3.4
## [228] bbmle_1.0.24
## [229] systemPipeR_1.24.3
## [230] boot_1.3-28
## [231] readr_2.1.4
## [232] geneplotter_1.68.0
## [233] ggplotify_0.1.0
## [234] Category_2.56.0
## [235] DEoptimR_1.0-11
## [236] bit_4.0.4
## [237] scatterpie_0.1.7
## [238] jpeg_0.1-8.1
## [239] ggraph_2.0.5
## [240] pkgconfig_2.0.3
## [241] rstatix_0.7.0
## [242] knitr_1.37