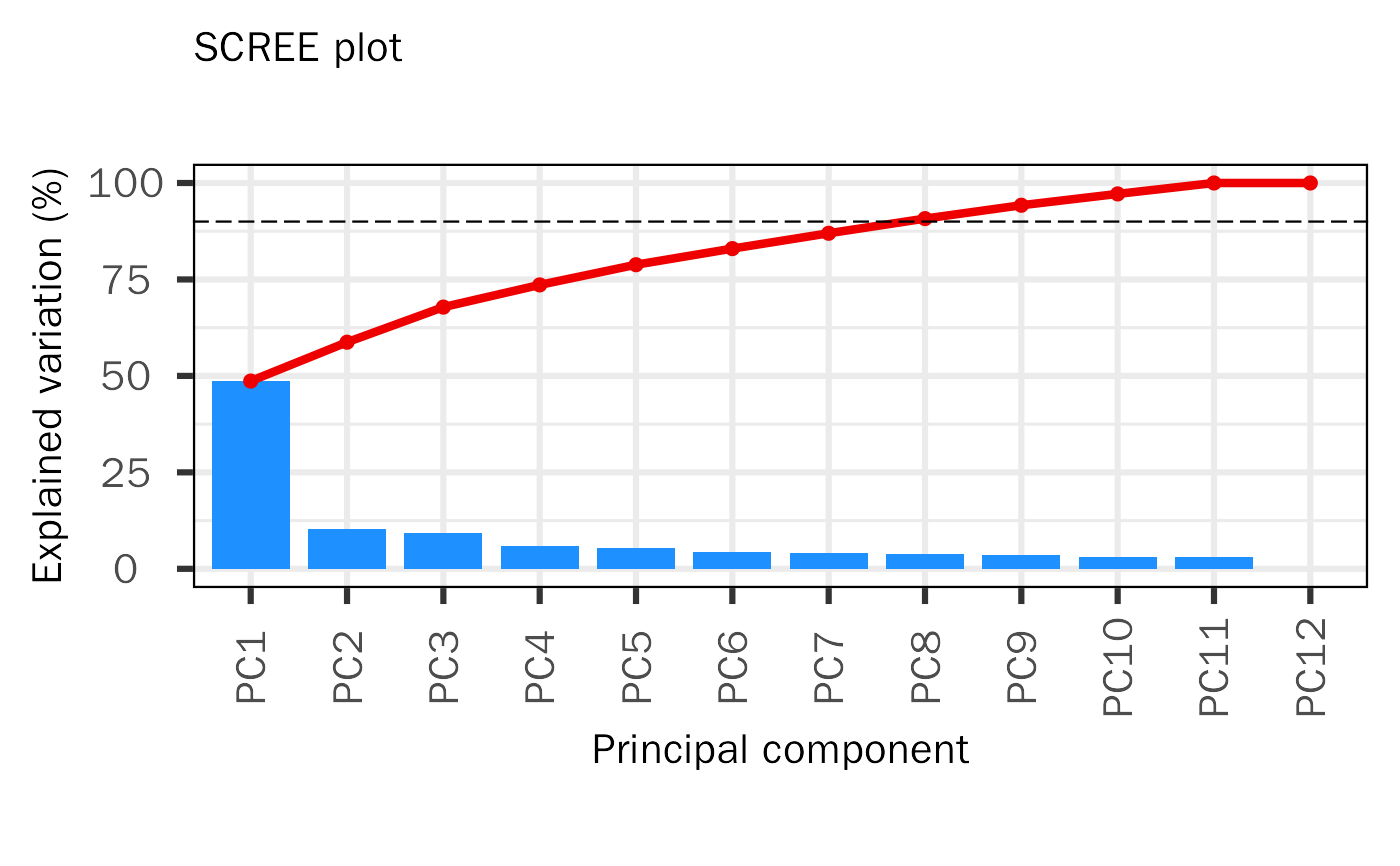
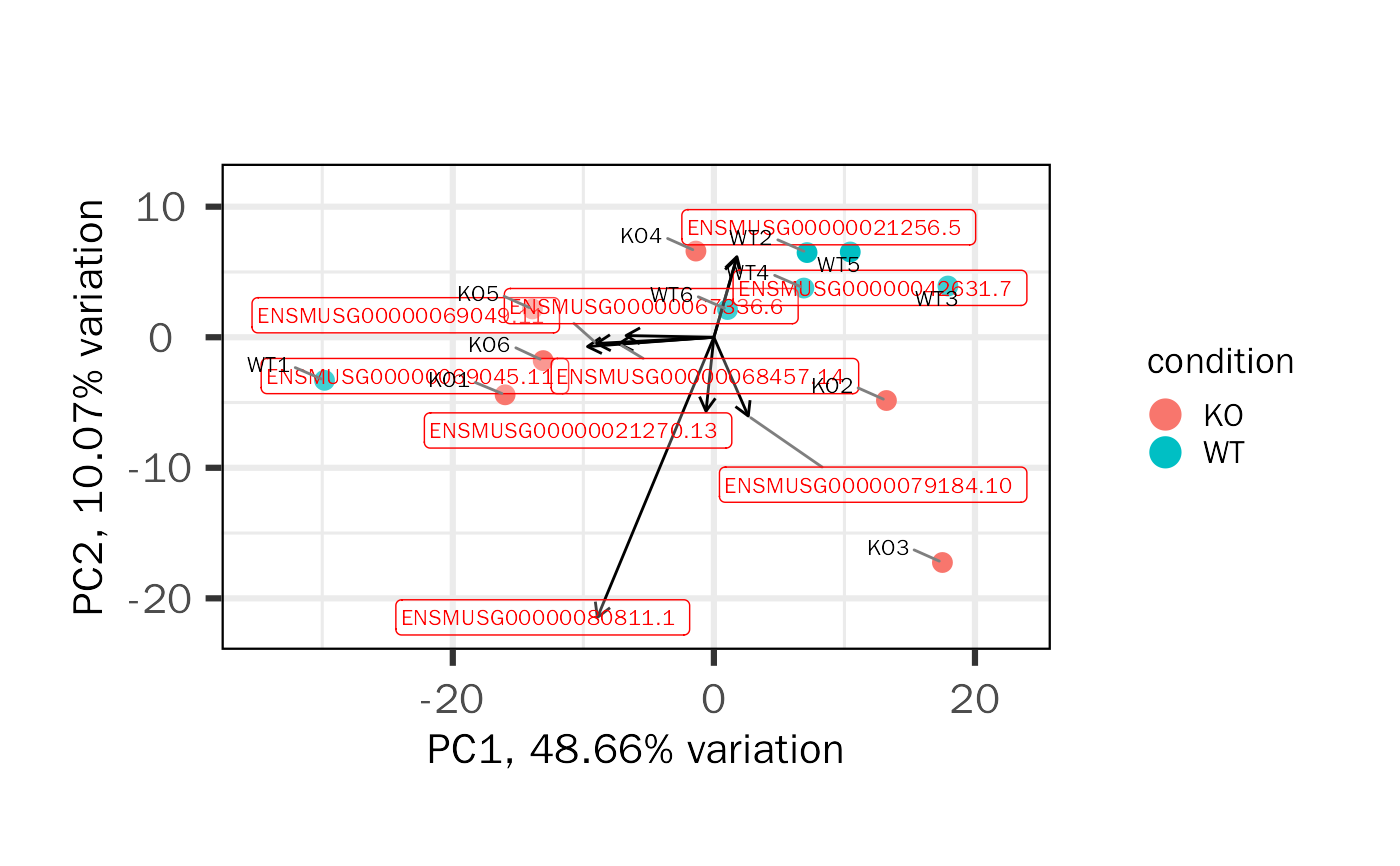
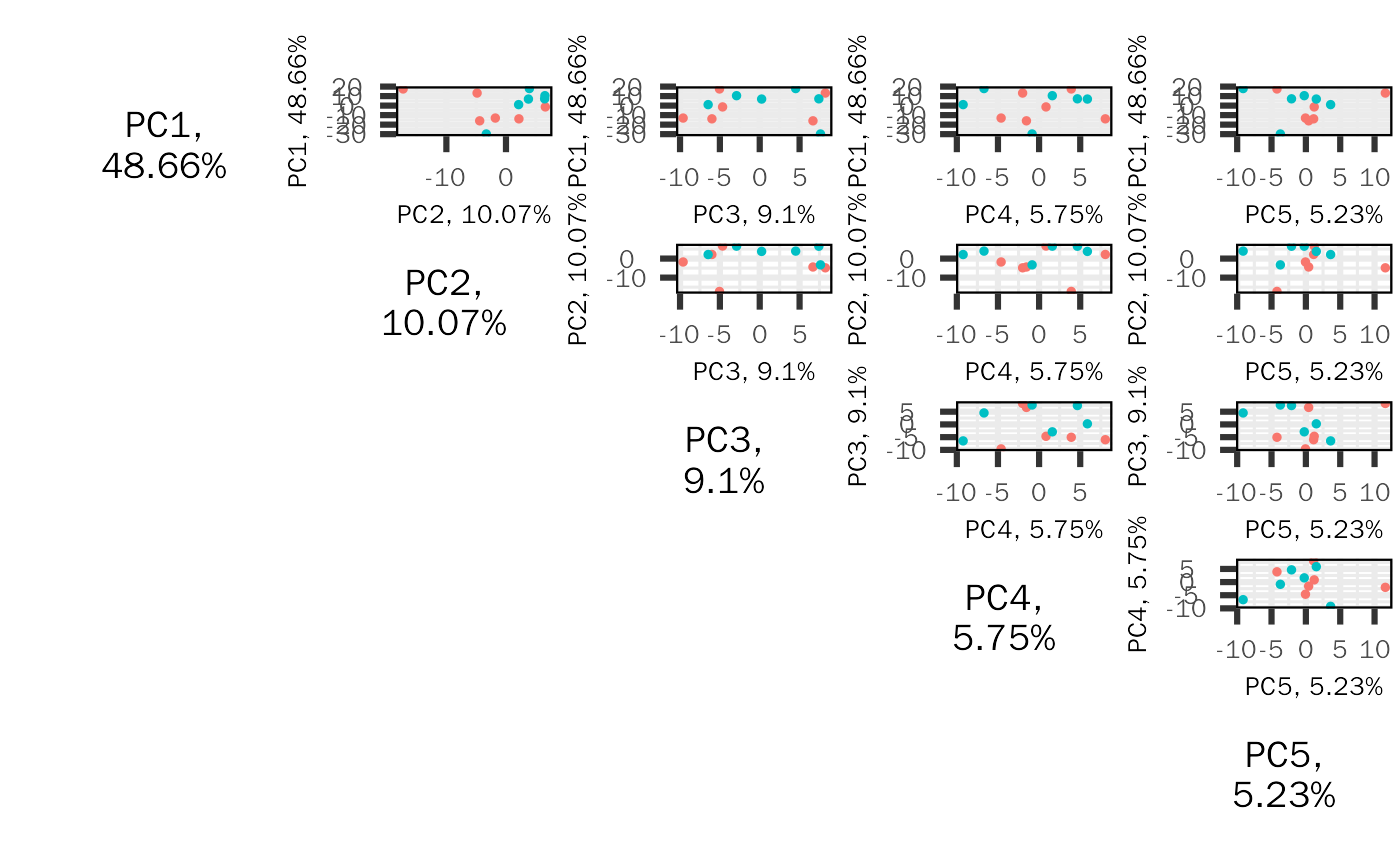
Generated PCA baisc plots, including screen plot, biplot and pairs plot.
PCABasic.RdGenerated PCA baisc plots, including screen plot, biplot and pairs plot.
PCABasic( pca, x = "PC1", y = "PC2", explain.threshold = 90, loading.num = 5, loading.label.size = 3, loading.label.color = "red", colby = NULL, pair.pc = 5, pair.label.size = 14, legend.pos = c("right", "bottom", "top", "left", "none") )
Arguments
| pca | PCA results of |
|---|---|
| x | The principal component to display on the x axis. Default: PC1. |
| y | The principal component to display on the y axis. Default: PC2. |
| explain.threshold | The threshold of explained variance. Default: 90. |
| loading.num | Select loading gene number based on absolute ordered variable loading for each PC in the biplot. Default: 5. |
| loading.label.size | Size of loading label. Default: 3. |
| loading.label.color | Color of loading label. Default: red. |
| colby | Group information to color samples. Default: NULL. |
| pair.pc | The principal components to be included in the plot. Default: 5. |
| pair.label.size | Size of p rincipal component label. Default: 14. |
| legend.pos | Position of legend ('top', 'bottom', 'left', 'right', 'none'). Default: right. |
Value
A ggplot2 object.
Examples
library(DESeq2) library(DEbPeak) count.file <- system.file("extdata", "snon_count.txt", package = "DEbPeak") meta.file <- system.file("extdata", "snon_meta.txt", package = "DEbPeak") count.matrix <- read.table(file = count.file, header = TRUE, sep = "\t") meta.info <- read.table(file = meta.file, header = TRUE) dds <- DESeq2::DESeqDataSetFromMatrix(countData = count.matrix, colData = meta.info, design = ~condition)#> Warning: some variables in design formula are characters, converting to factorskeep.genes <- rowSums(DESeq2::counts(dds, normalized = FALSE)) >= 10 dds <- dds[keep.genes, ] pca_res <- PCA(deobj = dds, transform.method = "rlog")#>#>PCABasic(pca_res, colby = "condition")#> #>#> #>#> #>#> #>#> #>#> #>#> #>#> #>#> #>#> #>#> #>#> #>#> #>#> #>#> #>#> $screen#> #> $biplot#> #> $pairs#>